Protein Quantification
iTRAQ
iTRAQ technology utilizes isobaric reagents to label the primary amines of peptides and proteins. The iTRAQ reagents usually consist of an N-methyl piperazine reporter group, a balance group, and an N-hydroxy succinimide ester group that is reactive with the primary amines of peptides. The balance groups present in each of the iTRAQ reagents function to make the labeled peptides from each sample isobaric and the quantification is facilitated through analysis of reporter groups that are generated upon fragmentation in the mass spectrometer. There are currently two mainly used reagents: 4-plex and 8-plex, which can be used to label all peptides from different samples/treatments. These samples are then pooled and usually fractionated by nano liquid chromatography and analyzed by tandem mass spectrometry (MS/MS).
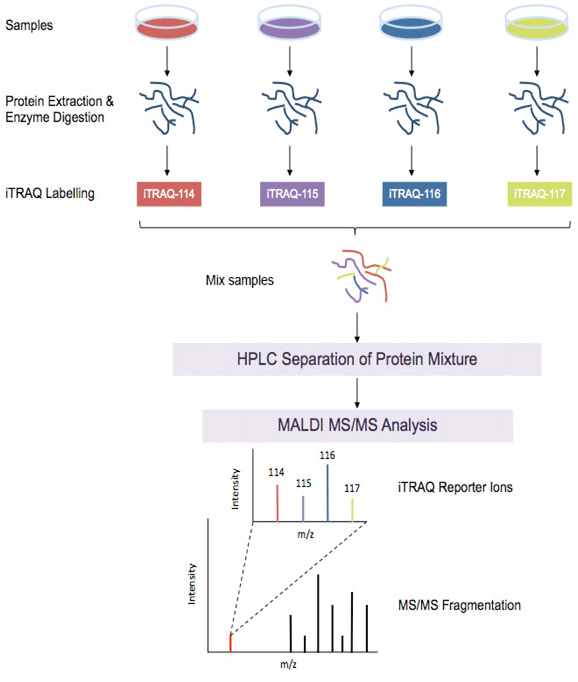
iTRAQ Workflow:
iTRAQ workflow (4-plex) is shown above. Samples to be quantified are prepared under various treatment conditions followed by cell lysis to extract proteins. After using a standard protein assay to estimate the protein concentration of each sample, proteins are digested using an enzyme, such as trypsin, to generate proteolytic peptides. Each peptide digest is labeled with a different iTRAQ reagent and then the labeled digests are combined into one sample mixture. The combined peptide mixture is analyzed by LC-MS/MS for both identification and quantification.
A database search is then performed using the fragmentation data to identify the labeled peptides and hence the corresponding proteins. The fragmentation of the attached tag generates a low molecular mass reporter ion that can be used to relatively quantify the peptides and the proteins from which they originated.
Advantage of our iTRAQ technique:
- Cutting-edge facilities & optimized protocols
- High sensitivity
- Untargeted approach for biomarker discovery
- Post-translational modification is detectable
TMT Based Proteomics Services
Tandem Mass Tag (TMT) method was designed by Thermos Fisher Scientific for the identification and quantification of proteins in different types of samples. Each TMT tagging reagent is composed of an amine-reactive NHS-ester group, a spacer arm and an MS/MS reporter. And the isobaric tagging reagent within a set has the same nominal parent (precursor) mass. These reporter ions are in the low mass region of the MS/MS spectrum and are used to report expression changes in cell-based or tissue samples that may not be amenable to metabolic isotopic labeling strategies such as SILAC.
The isobaric labels TMT (tandem mass tagging) are available in up to 11 tags that can be used for labeling practically any peptide or protein sample. TMT makes it possible to multiplex the analysis, to efficiently use the instrument time and exert further controls for technical variation. Due to its ability to multiplex up to 11 samples, TMT are widely used for quantitative protein biomarker discovery. TMTzero, TMTduplex, TMTsixplex, TMT10plex, and TMT11plex Reagents share an identical structure, allowing efficient transition from methods development to multiplex quantitation.
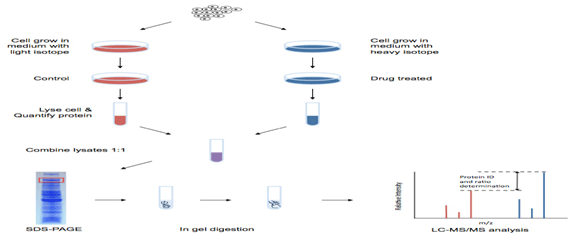
SILAC-based Proteomics Analysis
At Novelgene, we can provide a complete SILAC service, including cell culture, treatment of cells, and proteomics analysis. Based on SLIAC and mass spectrometry, we can analyze the relative proteomic change under differential treatments. We can offer of multiplex two or three samples per analysis which allows for increased throughput and cost savings in quantitative proteomics experiments along with improved relative quantitation.
Workflow of SILAC-based Proteomics Analysis
- Cell culture and cell labeling.
- Sample preparation, including protein extraction, mixing unlabeled (light) and labeled (heavy) samples, trypsin digestion, peptide fractionation.
- LC-MS/MS analysis. An advanced platform equipped Triple TOF 5600, Q-Exactive, Orbitrap Fusion™ Tribrid™, etc.
- Data analysis by using professional software and database for reliable results.
- Detailed report.
Sample Requirement
- We can accept not only non-labeled cell samples, but also samples at any stage after SILAC labelling.
- Non-labeled cell samples
- Labeled cell samples
- Protein samples
- If you want to know specific samples requirements, please feel free to contact us.
Advantages of SILAC-based Proteomics Analysis Service
- High labeling rate: The labeling efficiency is not affected by lysate and efficiency can be as high as 100%.
- High sensitivity: The sample requirements are small, usually only tens of micrograms of protein per sample.
- High throughput: Mass spectrometry can identify and quantify hundreds to thousands of proteins simultaneously.
- High precision: Multiple samples are mixed, simultaneously digested, and identified. The subsequent experiments have the same effect on the sample, which reduces the impact of experimental operation and equipment, and has higher precision and reproducibility.
Absolute Quantification (AQUA)
There are two stage of this method, 1st stage is: a representative tryptic peptide is selected and standard synthesized from the protein of interest with an amino acid sequence that exactly mimics the corresponding native peptide produced during proteolysis. And stable isotopes are incorporated during the synthesis process. Then, the synthetic AQUA internal standard peptide is next evaluated by LC-MS/MS. The SRM analysis is then performed to measure a specific precursor-to-product ion transition. and the second stage of the AQUA strategy is its implementation to measure the amount of proteins or modified proteins in a complex mixture. Cell lysates can be optionally separated prior to proteolysis to increase the dynamic range of the assay. We have most commonly used SDS-PAGE for this enrichment, followed by excision of the region of the gel where the protein of interest migrates. In-gel digestion is performed in the presence of the AQUA peptide, which is added to the gel pieces during the digestion process. Following proteolysis, the complex peptide mixture, containing both heavy and light peptides, is analyzed in an LC-SRM experiment using parameters determined during Stage 1.
Benefits of Our AQUA Strategy
- Customized assay panel development and sample measurement
- Very accurate method (variation CV<5%)
- Robust and reproducible quantitation
Novelgene Technologies also provide the following bioinformatics services in Absolute Quantification:
- Functional annotation and enrichment analysis
- Clustering analysis
- Network analysis
- Statistical analysis
Label-free Quantification
Label-free quantification is a method of determining the relative amount of proteins in two or more biological samples, but unlike other quantitative methods, it does not use a stable isotope for chemical binding and labeling of proteins. Label-free quantitative proteomics approach provides a powerful tool to resolve and identify thousands of proteins from a complex biological sample. In this approach, proteins are first digested with a protease into a peptide mixture, which is subsequently analyzed by tandem MS (MS/MS) and identified by database searching. Relative protein abundance is determined by either spectral counting or chromatographic peak intensity measurements. Compared with other proteomic methods, the label-free quantification approach is rapid and more sensitive, which increases the protein dynamic range 3- to 4-fold as compared to 2DE. This method can also be automated with the ability for large-scale proteome analysis.
Deliverables
- Detailed reports including experimental procedures, instrument parameters, etc.
- Raw data and analysis results.
Benefits of Label-free Approach Working for Our Customers
- Simple experimental process
- No limit on the number of samples
- Suitable for multiple types of samples
- Excellent analytical reproducibility
Semi-quantitative Proteomics Analysis
In Proteomics Research, there are two main types of research analysis: QUALITATIVE and QUANTITATIVE. Between these two extremes is semi-quantitative analysis, which assigns approximate measurements to data, rather than an exact measurement. Often used in cases where a direct measurement is not possible, but inference is unacceptable. In particular, the analysis is useful in cases where quantified data might fluctuate periodically.